
Approach to Drug Therapy for Epilepsy
Robert S. Fisher, MD, PhD
Department of Neurology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
Medications remain the mainstay of treatment for epilepsy. About 75% of individuals with epilepsy can be controlled adequately with antiepileptic drugs (AEDs). The remainder continue to have seizures or unacceptable toxicity from medication. Clinicians should use AEDs according to the following principles: decide whether to treat patients with AEDs, select the proper drug for the seizure type, initiate therapy slowly, simplify the regimen to improve compliance, and determine the length of treatment. Therapy usually is initiated with phenytoin or carbamazepine for partial seizures and with valproic acid for primarily generalized seizures. Phenobarbital is a second-line medication because of its side effects. Felbamate, the first of the new wave of AEDs, is potentially toxic. Gabapentin is well-tolerated and useful as adjunctive therapy for partial seizures. Lamotrigine is indicated for partial seizures but has broad-spectrum efficacy and convenient dosing. Topiramate is an effective medication for partial seizures but may cause cognitive limitations. Tiagabine increases GABA at the synapse and appears useful for partial seizures. Other drugs are in various stages of development. New forms of older AEDs include parenteral valproic acid; fosphenytoin, a water-soluble form of phenytoin; rectal diazepam gel; and long-acting carbamazepine medications. The newer medications rarely eliminate seizures in previously intractable patients but may offer a better risk-benefit profile and a new approach for the large population of people with inadequately controlled seizures.
Key Words: carbamazepine, epilepsy, felbamate, gabapentin, lamotrigine, pharmacology, phenobarbital, phenytoin, seizures, tiagabine, topiramate, valproic acid, vigabatrin, zonisamide
Although other articles in this issue demonstrate the importance of surgical and nonpharmacological therapies, the mainstay of epilepsy treatment remains antiepileptic drug (AED) therapy. The goal of AED therapy should be complete seizure control with no or minimal toxicity. The definition of complete seizure control is elusive. In broad terms, however, it means sufficient control to allow patients to live their lifestyle without epilepsy-related limitations. Freedom from seizures for a year or a few years may suffice to achieve this goal. Approximately 75% of people with epilepsy can achieve satisfactory control of their seizures on medical therapy.13 The remaining 25% continue to have unacceptable seizures or toxicity and are candidates for surgery or alternative therapies. These individuals also need diagnostic reevaluations to confirm the epilepsy diagnosis.
The ideal medicine for epilepsy would be effective, safe, easily absorbed, and inexpensive. It would also have few side effects, favor compliance with a single daily dose, and lack drug interactions. No such medication exists, and seizure pharmacotherapy largely consists of balancing these presently disparate goals. This article reviews the characteristics and efficacy of recently developed AEDs as well as new forms of old AEDs.
Principles of Antiepileptic Drug Therapy
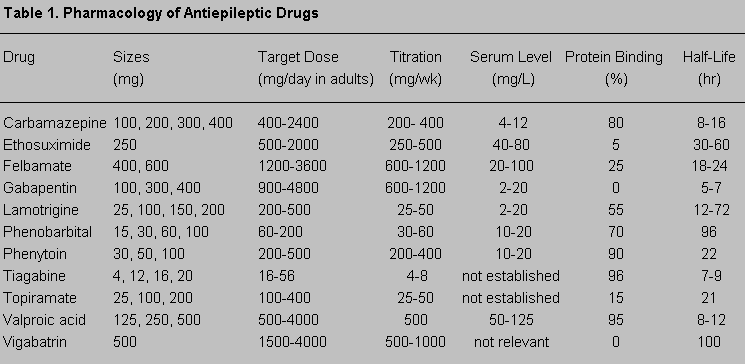
Clinicians should base the use of AEDs on several principles and knowledge of properties of individual drugs (Table 1). First, the decision should be made whether to treat the patient with an AED. Some seizures are so minor or so infrequent that drug treatment may not be justified. Second, clinicians should choose the best regimen for the drug (Fig. 1). According to results from the Veterans Administration (VA) Cooperative Study,51,52 carbamazepine and phenytoin are the drugs of choice for partial seizures, although justification exists for using other drugs as adjunctive or second-line therapy for partial seizures. Generalized seizures usually are treated with valproic acid or a benzodiazepine. Lamotrigine57 and topiramate9 are effective for many types of generalized seizures, but they have no official indication for generalized seizures in the United States. Myoclonic seizures are treated with valproic acid, benzodiazepine, lamotrigine, or zonisamide.26 Traditionally, absence seizures are treated with ethosuximide. Valproic acid is also effective and covers any associated tonic-clonic seizures.
There is latitude for clinicians to choose the drug of choice for epilepsy based upon their familiarity with the drug, side effects, ease of use, and cost. Clinicians should select a simple regimen to encourage compliance, one of the most important practical issues in the pharmacotherapy of epilepsy. Regimens of a single daily dose are far simpler than regimens that require four daily doses.20
Serum levels of AEDs can be used selectively to verify compliance, to help determine idiosyncratic toxicity, or to identify the likely toxic medication when patients are taking multiple medications. When possible, clinicians should prescribe one rather than two or three AEDs.30 The VA Cooperative Study51 suggested that only 10% of patients benefit significantly from the addition of a second medication. In seizure clinics enriched with populations of intractable patients, this number may be higher. Additionally, the transformation from a regimen with three or four drugs to a monotherapy regimen can be extremely difficult because of withdrawal seizures. Monotherapy is a worthy goal but often unachievable in practice.
Epilepsy therapy is not necessarily a life sentence. If patients are free of seizures for 2 to 5 years, have a normal electroencephalogram (EEG), and no continuing predisposition (e.g., juvenile myoclonic epilepsy or brain lesions), have not been in status, and are willing to take a chance, they may be candidates for drug withdrawal. An EEG with epileptiform discharges serves to discourage withdrawal.2 Approximately one of each three patients tapered from seizure medicines in these circumstances will relapse.16 I ask patients to refrain from driving for 3 months from the start of the medication taper to avoid a withdrawal seizure behind the wheel. Some patients are discouraged from tapering because of the driving issue and the 33% risk of withdrawal seizures. In these instances, it is usually preferable to continue patients on seizure medications.

New Medications
The first effective drug for epilepsy was bromides,32 but bromides were too toxic for routine use (Fig. 2). Phenobarbital, introduced in 1912 as a sedative,85 was soon found to have antiepileptic properties. Other barbiturates and compounds with similar structures were developed by imitation and trial and error based upon the phenobarbital molecule. Phenytoin (Dilantin® ) was introduced in 193854 after a discovery based on newly developed laboratory screening model systems. Subsequently, ethosuximide was released for absence seizures,25 carbamazepine for partial seizures,51 and valproate52 for generalized and, later, partial seizures. After the release of valproate in the United States in 1978, a 15-year gap ensued and no major new drug was released. Then, between 1993 and 1998, five new medicines were released (Fig. 3): felbamate,11 gabapentin,81 lamotrigine,50 topiramate,28 and tiagabine.67 The release of two others, zonisamide45 and possibly vigabatrin,31 appears imminent. Several others have been tested in clinical trials and are targeted for release over the next few years.10
Felbamate
The first new medication released after valproate was felbamate. Felbamate (Felbatol®, Carter-Wallace, Cranbury, NJ), an effective medication with both desirable and undesirable properties, is an analogue of the meprobamate receptor.12 It is useful for treating partial seizures, atonic seizures, and probably a broad spectrum of other seizures as well. Felbamate induces weight loss rather than weight gain and tends to increase rather than decrease alertness. Side effects usually consist of nausea, headache, insomnia, behavioral disturbances in children and less often adults, and a wide variety of drug interactions.
Because of its potential for inducing aplastic anemia and hepatotoxicity during the first few years of use, felbamate is the subject of a “black box” warning from the Food and Drug Administration (FDA).61 Recent experience has suggested that aplastic anemia from felbamate is rare—31 cases have been reported to the manufacturer. In the past several years no cases have been reported.42 Patients with prior immunological, hematological, or systemic disease may be at higher risk for complications.
Felbamate is available as 400 and 600 mg pills and typically is given in doses of 1800 to 3600 mg/day divided between two doses. The drug, however, is now used as a last resort because of the associated risks of aplastic anemia and liver toxicity.
Gabapentin
Gabapentin (Neurontin®, Parke-Davis, Morris Plans, NJ) was the second new drug for epilepsy in the recent wave of AED introductions. Gabapentin was developed as an analogue of gamma-aminobutyric acid (GABA, the main inhibitory neurotransmitter in the brain) with a cyclohexane ring attached to increase its penetration through the blood-brain barrier.18 Despite its similarity to GABA, the drug does not bind to GABA receptors. Nor does it increase synthesis of GABA, decrease GABA degradation, or raise extracellular levels of GABA. Operating through a novel uptake site,40 however, gabapentin may increase intracellular neuronal GABA. Gabapentin also inhibits calcium currents.77
Gabapentin is indicated for adjunctive therapy of partial seizures. Monotherapy trials have been completed,7 and a monotherapy indication is under consideration by the FDA. Gabapentin does not appear useful for absence or myoclonic forms of epilepsy. Pivotal trials of gabapentin 3,81,82 were performed at daily doses of 900 to 1800 mg. Treating clinicians, however, often must increase the daily dose to 2400, 3600 or 4800 mg to achieve full efficacy. At high doses, absorption becomes limited by a saturable transport protein. Gabapentin comes as 100, 300, and 400 mg capsules. Because of its half-life of 5 to 7 hours, gabapentin should be taken three times a day. Essentially, gabapentin is excreted unchanged via the kidney and therefore avoids hepatic metabolism. Nonhepatic metabolism, a relative lack of drug interactions, and the near absence of protein binding render gabapentin a convenient drug to use with other medications.
Gabapentin efficacy can be reported in terms of responder rate—the percentage of people with a 50% or greater reduction in seizures. Controlled clinical trials document the gabapentin responder rate to be 20 to 30%.3,81,82 Open label trials conducted with individuals less affected than those who participate in pivotal trials and with higher doses of medication have shown higher levels of efficacy. Adverse events consist of somnolence, dizziness, blurred vision, ataxia, fatigue, and other neurologically based, reversible symptoms. Overall, gabapentin is very well tolerated. In the United States and Britain,81,82 only 2 to 6% of patients withdrew from clinical trials because of severe adverse events attributable to gabapentin.
Typically, gabapentin is initiated with 300 or 400 mg pills, and the dose is increased each day to 900 to 1200 mg. During the next few weeks, the medication can be increased to a maximum dose of 4800 mg. Other concurrent medications seldom require adjustment. The ability to titrate the medication rapidly is an advantage of gabapentin.
Lamotrigine
Lamotrigine (Lamictal®, Glaxo Wellcome, Research Triangle Park, NC) is an analogue of folate antagonists but itself has little antifolate action.68 The drug appears to work by blocking sodium channels,19 by inhibiting rapid neuronal firing, and by inhibiting the release of the excitatory neurotransmitter, glutamate. Lamotrigine is approved as an add-on drug and is useful for treating partial seizures. Clinical trials, however, have suggested a broad spectrum of efficacy along with utility in generalized seizure disorders.57
The half-life of lamotrigine depends upon concurrent medications. In monotherapy it has a half-life of about 24 hours and can be taken as a single daily dose. With hepatic inducers, such as phenytoin, phenobarbital, or carbamazepine, its half-life falls to 12 hours and dosing twice daily is recommended. Valproic acid inhibits hepatic metabolism and increases the half-life and serum level of lamotrigine several fold. Concurrent medications must therefore be considered when a lamotrigine regimen is planned.
The responder efficacy rate of lamotrigine in partial seizures is similar to that of gabapentin. In trials of intractable epilepsy, the frequency of seizures is at least halved in 20 to 30% of patients.8,41,46,50,56,72 Common side effects of lamotrigine result from central nervous system (CNS) toxicity: dizziness, headache, diplopia, ataxia, nausea, blurred vision, or somnolence. The side effect of main concern is rash, which occurs in 5 to 10% of patients on the medication.55 Rash is usually benign and transient, but lamotrigine has provoked a few serious cases of Stevens-Johnson syndrome or toxic epidermal neurolysis. The risk for rash appears to be higher in pediatric patients and in patients simultaneously taking valproic acid. Slow titration reduces the risk for rash. The recommended target dosage for adults ranges between 300 and 500 mg, but some individuals benefit from higher doses. Lamotrigine is available in 25, 100, 150, and 200 mg pills. In our clinic we start medication at 25 mg/day and increase the dose by 25 mg each week on a twice daily (b.i.d.) regimen to 100 mg b.i.d. At that point in the typical adult, we switch to 100 mg pills and increase to 200 mg b.i.d. over the next few weeks.
Topiramate
Topiramate (Topamax®, Ortho-McNeil, Raritan, NJ) is an AED developed in conjunction with screening programs of the Epilepsy Branch of the National Institutes of Health.27 It exhibits multiple potentially useful AED mechanisms, including inhibition of sodium channels, inhibition of the glutamate AMPA receptor, potentiation of GABA neurotransmission, and weak inhibition of carbonic anhydrase.53 Topiramate is well absorbed after an oral dose with or without food, binds approximately 15% to proteins, and is excreted by the liver and kidney. It has a half-life of approximately 21 hours requiring twice daily dosing. Phenytoin and carbamazepine reduce topiramate serum levels by about 50%. Topiramate, in turn, can increase levels of phenytoin.
As with most new AEDs in the United States, topiramate is licensed as adjunctive therapy for partial seizures. In fact, topiramate may have more broad spectrum activity, also benefitting generalized tonic-clonic, myoclonic, absence, and atonic seizures.5 The efficacy of topiramate in clinical trials has been good, with a 40 to 50% responder rate when the drug is added to the regimens of patients with intractable partial epilepsy.5,28,62,76,79 For most adults, its efficacy is near maximum at 400 mg/day,4 after which further increases enhance drug toxicity.
The usual CNS reversible side effects of somnolence, dizziness, ataxia, and diplopia can occur with topiramate therapy.27 Renal stones have been reported in 1 to 2%. Approximately 30% of people on topiramate develop some type of cognitive difficulty: slowness of thinking, confusion, amnesia, and word-finding difficulties. Some of these changes are best noted by the patient’s relatives rather than by the patient him- or herself. The cognitive side effects can be minimized by slow-dosage titration.
I use the same titration schedule as for lamotrigine, starting at 25 mg/day. Each week the dose is increased by 25 mg/day b.i.d. until 100 mg/day b.i.d. is reached. At that point, I switch patients from the 25 mg to 100 mg pill and increase dosage over the next few weeks to 200 mg b.i.d. Some patients achieve efficacy with topiramate at 50 to 200 mg/day. Phenytoin doses may need to be reduced by about a third because of a drug interaction.
Tiagabine
Tiagabine (Gabitril®, Abbott & Novo Nordisk, North Chicago, IL and Princeton, NJ) is a drug that emerged from our understanding of AED mechanisms.35 Tiagabine is designed to inhibit the uptake of GABA into the presynaptic nerve terminal, thereby leaving more GABA at the synapse for inhibition. So far, no other major mechanism of action has been observed for tiagabine.
Tiagabine is available in 4, 12, and 16 mg pills, and typical daily doses range from 16 to 56 mg. Therapy is initiated with 2 mg pills and increased 2 mg every 1 or 2 weeks until a target dose of 16 to 56 mg/day b.i.d. has been reached. Even though tiagabine has a half-life of 6 to 8 hours, suggesting the need for dosing three or four times a day, a clinical trial showed no loss of efficacy when doses were administered twice compared to four times a day.
Tiagabine is indicated as adjunctive treatment for partial and secondarily generalized seizures for patients 12 years and older. The drug is metabolized by the hepatic enzyme system. At 56 mg/day, the responder rate ranges between 20 and 30% for partial seizures and is as high as 40 to 50% for secondarily generalized seizures.65,67,83 Monotherapy studies have been completed,70 and a monotherapy indication is pending.
The most frequently encountered adverse events with tiagabine are dizziness, fatigue, nervousness, tremor, and abnormal thinking (cognitive slowing).67 As with several other GABA-related drugs, tiagabine occasionally produces psychiatric side effects. The incidence of psychosis, however, is similar to that of placebo-treated patients.1 Tiagabine can produce paradoxical worsening of seizures, particularly absence seizures.71
Vigabatrin
Vigabatrin (Sabril®, Hoechst Marion Roussel, Kansas City, MO) is a drug designed to increase levels of GABA in the brain by inhibiting GABA-transaminase, the main enzyme that metabolizes GABA.73 Vigabatrin is available in most major countries of the world but not in the United States at the time of this writing.
Initially, concerns were raised about animal toxicology findings in mice, rats and dogs, which showed dose- and time-dependent swelling of myelin.15 Such changes, which are visible on magnetic resonance (MR) imaging, are reversible when use of the drugs is discontinued. Furthermore, the changes have not been shown to occur convincingly in humans, with hundreds of thousands of patient-years world experience (Personal communication, Hoechst, Marion, Roussel, 1999). As with other drugs that increase the levels or function of brain GABA, psychiatric complications in the form of depression and occasional psychosis have also been observed.29 A recent serious concern has arisen over concentric or patchy visual field defects from vigabatrin, the incidence and mechanism of which are unknown.38,44 The FDA has expressed concern about visual field problems resulting from vigabratin, and this may impede the drug’s release in the United States.
Despite these concerns, vigabatrin is sometimes a particularly efficacious drug for intractable epilepsy. Multiple studies show responder rates of 30 to 50%.6,31 Vigabatrin is also useful in patients with infantile spasms associated with tuberous sclerosis and in those with atonic seizures.84
Vigabatrin is metabolized renally, which simplifies consideration of its use together with hepatically metabolized medications. It inhibits GABA transaminase for as long as 5 days, rendering serum levels irrelevant. Protein binding is negligible. Interactions with other AEDs are minimal, although a lowering of phenytoin levels by about 20 to 33% has been observed.66 Vigabatrin is available as 500 mg pills, and the typical dose ranges between 1500 and 3000 mg/day b.i.d. Experience around the world and in clinical protocols suggests that vigabatrin can be initiated at a dose of 500 mg/day and then increased by 500 mg/week to full dose or tolerance. Attention should be paid to psychiatric difficulties and to any changes in vision or visual fields.
Zonisamide
Zonisamide (Zonegran®, Dainippon, Athena-Elan, South San Francisco, CA) is a sulfonamide-related drug.75 It was originally studied in the United States,45 but in response to the induction of a few renal stones drug trials were put on hold. Further trials in Europe74 and Japan75 suggested that the risk for renal stones was tolerable, and the drug has subsequently completed pivotal trials in the United States. At the time of this writing, zonisamide has been issued a letter of approvability by the FDA and is expected to be available within the year.
Zonisamide is available as 100 mg tablets, and the typical dose is 400 mg/day. As with topiramate, higher doses seem to add cognitive problems without increasing efficacy. Zonisamide has a half-life of 30 to 60 hours, enabling single daily dosing or b.i.d. dosing as desired. It does not appear to have major interactions with other AEDs. Zonisamide can be teratogenic in other species and perhaps also in humans.43 In human trials, cognitive difficulties and renal stones have occurred as side effects. Zonisamide may be particularly effective in myoclonic epilepsies.39 Recently completed randomized trials of zonisamide have not yet been published.
Oxcarbazepine
Oxcarbazepine (Trileptal®, Novartis, Summit, NJ) has a keto group attached to the 10 position of the carbamazepine three-ringed structure.22 This oxygen molecule inhibits metabolism to the carbamazepine 10,11-epoxide, which may account for some of the side effects of carbamazepine. Therefore, the efficacy of oxcarbazepine may be found to be similar to that of carbamazepine, but its therapeutic ratio may be more favorable.33 One randomized study showed oxcarbazepine to be equivalent to phenytoin in efficacy against partial seizures but better tolerated.36 The active compound is the monohydroxy derivative of oxcarbazepine, which is being tested in intravenous form as an AED in its own right. In the United States, oxcarbazepine is expected to be released in 1999 or 2000.
Ganaxolone
Ganaxolone (CoCensys, Irvine, CA) is a neurosteroid related to the hormone progesterone.58 Ganaxolone, however, has little hormonal action and appears to inhibit seizures by modulating the GABA receptor at its neurosteroid site.17 Single doses to 1500 mg for one dose, and chronic multiple doses to 300 mg b.i.d. have been tolerated well by volunteers.58 A clinical trial of ganaxolone showed a benefit compared to a placebo at the p = .07 level, and the drug is being considered for further development.
Remacemide
Remacemide (Astra Merck, Wayne, PA) is a noncompetitive, weakly binding, N-methyl-D-aspartate (NMDA) antagonist.23,59 A drug capable of inhibiting seizures by blocking the NMDA subtype of the glutamate receptor has long been sought, but psychotropic side effects have limited the use of such medications. Remacemide may be relatively free from psychotropic side effects, perhaps because of its weak binding to the NMDA receptor78 or because of its additional action against sodium channels. Remacemide has been given in doses ranging from 400 to 800 mg/day divided either twice or four times daily. Its metabolite, the desglycinyl compound, has a half-life of 8 to 15 hours and is 90% protein bound. Remacemide has significant drug interactions, increasing serum levels of phenytoin and carbamazepine in patients taking these drugs concurrently. Gastrointestinal upset, dizziness, and mood changes have been seen in early clinical trials. If it proves safe and effective in treating epilepsy, remacemide would be a useful addition to our available seizure medicines because it would enable the blockade of excitatory neurotransmission during seizures. Such evidence awaits completion of clinical trials.
Levetiracetam
Levetiracetam (ucb LO59, UCB Pharma, Smyrna, GA) is related to the nootropics, of which the best known member is piracetam. The drug has shown efficacy in animal models and a promising therapeutic ratio in early human trials.47 Pivotal trials have been completed and hope is for release in the near future.
Loreclezole
Loreclezole (Janssen, Titusville, NJ) is a triazole derivative with a profile in animal models of epilepsy similar to that of barbiturates. It interacts with the GABA receptor.69 It also increases the levels of extracellular serotonin.21 Its half-life is long at 10 to 30 days. A randomized trial of loreclezole in 62 patients with partial seizures showed a 50% or greater reduction in seizures in 19% of the patients.64
Rufinamide
Rufinamide (Novartis, Summit, NJ) is a compound developed in Europe. It has a novel structure, unrelated to other AEDs. Preliminary European trials suggest that it has a broad therapeutic ratio, and the drug is now in clinical trials in the United States.60 Expectations are that it will be efficacious for partial and secondarily generalized seizures.
SKF 204269
SKF 204269 (SmithKline Beecham, Pittsburgh, PA) interacts with potassium channels on neurons to inhibit excessive neuronal firing. This drug has completed randomized clinical trials for adjunctive therapy of partial seizures, and a new drug application is under preparation.
Pregabalin
Pregabalin (Parke-Davis, Morris Plains, NJ) is related to the drug gabapentin, over which it may have certain benefits including a better therapeutic ratio and a longer half-life. It is entering clinical trials in the United States.
New Forms of Old AEDs
Several new forms of old AEDs are now also available. For example, Carbamazepine Extended Release (Tegretol-XR® , Novartis, Summit, NJ)80 is available as osmotically released capsules and as differentially released triple-time capsules (Carbatrol® , Athena, Elam, South San Francisco, CA).34 An injectable preparation of carbamazepine is not yet available in the United States.
Parenteral valproic acid (Depacon®, Abbott, North Chicago, IL) is available for intravenous use24 when oral valproic therapy cannot be continued. Its use in status epilepticus is not currently an approved indication because studies for status epilepticus have not been completed. Dosage of its intravenous form is the same as that of its oral form. Local tissue irritation at the intravenous site, unique to the intravenous formulation, is the main adverse event.
Fosphenytoin (Cerebyx®, Parke-Davis, Morris Plains, NJ) is a phosphonated form of phenytoin, more soluble and less irritating to tissues than phenytoin.14,48 Fosphenytoin is only for intravenous use. Each 1.5 mg of fosphenytoin is cleaved by endogenous phosphatases to 1 mg of phenytoin, but fosphenytoin is labeled as phenytoin equivalents (PE), such that 1000 mg of fosphenytoin (PE) produces 1000 mg of phenytoin. Fosphenytoin can be injected intravenously at a rate of 150 mg/min compared to a maximum of 50 mg/min for phenytoin. The ability to give fosphenytoin faster allows therapeutic serum levels to be established as quickly as can be obtained with phenytoin. Additionally, fosphenytoin can be given by intramuscular injection.63 Absence of the propylene glycol vehicle required to dissolve intravenous phenytoin may reduce the incidence of hypotension and cardiac arrhythmias associated with fosphenytoin; however, evidence supporting this claim awaits more experience.
Selecting a New Antiepileptic Medication
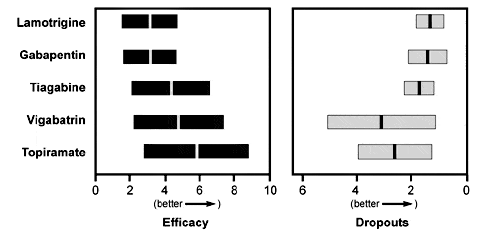
Available data on AEDs do not support a dogmatic approach to the introduction of new drugs. No single old or new AED stands above all the others in terms of its benefit or absence of toxicity. Treating physicians have both the opportunity and the need to use clinical judgment and experience when selecting a new seizure medicine. As a general rule, the more efficacious medications are more poorly tolerated (Fig. 4).49 It is unknown whether this trend reflects intrinsic differences among the new medications or simply different points along the dose-response curve. No large, randomized, controlled clinical trial of new drugs matched against each other or against the traditional AEDs has yet been completed.
Uncontrolled clinical experience suggests that some of the new drugs are believed to have better therapeutic ratios and possibly fewer side effects than the older medications. Several medications, such as gabapentin and vigabatrin, have fewer drug interactions than older medications whereas felbamate has more. The newer AEDs probably require fewer tests of their blood levels and routine blood tests, although blood parameters should still be monitored as indicated clinically. Recently introduced AEDs are not based upon the barbiturate structure and therefore are useful in patients who have multiple allergies to AEDs. Some of the drugs have different mechanisms of action. The primary advantage of the new medications is that they provide an opportunity to try something new in the very needy population of individuals with uncontrolled seizures.
The addition of one of the newer antiepileptic medications rarely renders a previously intractable patient free of seizures. Therefore, use of the new drugs should usually be considered palliative or an attempt to reduce side effects. Such a change in medication can improve the quality of life of individuals with epilepsy. At present, there is no easy way to predict which patients will respond best to which old or new AEDs. A logical course of brief drug trials, one after another, is therefore a rational approach. Nevertheless, such a trial should not be continued for years without success. Should a patient fail several of the old and new drugs, we consider the patient for curative epilepsy surgery (see Surgical Treatment of Intractable Epilepsy by Smith et al. on p. 27 of this issue) or palliative therapy with vagus nerve stimulation.37
The past 5 years have been exciting for epilepsy specialists because of the prolific development of new AEDs. Recent advances in neurosciences and molecular biology may next bring us drugs that can repair the underlying genetic defects in epilepsy. Such therapy would move our capabilities beyond seizure suppression into the area of true AED therapy.
Acknowledgments
The author was supported by the Sandra Solheim Aiken Fund for Epilepsy, the Newsome Chair for Neurology and the Women’s Board of the Barrow Neurological Foundation. The secretarial help of Ms. Beverly Moore and Ms. Jennifer Dunbar is gratefully acknowledged.
Disclosure
The author is on the speaker’s bureau, research panel, or consultant’s panel for several companies that make commercial products discussed in this review, including Parke-Davis, Glaxo-Wellcome, Ortho-McNeil, Abbott Pharmaceuticals, and Novartis. The author does not hold an equity position in any of the above companies.
References
- Adkins JC, Noble S: Tiagabine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the management of epilepsy. Drugs 55:437-460, 1998
- Andersson T, Braathen G, Persson A, et al: A comparison between one and three years of treatment in uncomplicated childhood epilepsy: A prospective study. II. The EEG as predictor of outcome after withdrawal of treatment. Epilepsia 38:225-232, 1997
- Anhut H, Ashman P, Fuerstein TJ, et al: Gabapentin (Neurontin) as add-on therapy in patients with partial seizures: A double-blind, placebo-controlled study. The International Gabapentin Study Group. Epilepsia 35:795-801, 1994
- Ben-Menachem E: Clinical efficacy of topiramate as add-on therapy in refractory partial epilepsy: The European experience. Epilepsia 38:S28-S30, 1997
- Ben-Menachem E, Henriksen O, Dam M, et al: Double-blind, placebo-controlled trial of topiramate as add-on therapy in patients with refractory partial seizures. Epilepsia 37:539-543, 1996
- Beran RG, Berkovic SF, Buchanan N, et al: A double-blind, placebo-controlled crossover study of vigabatrin 2 g/day and 3 g/day in uncontrolled partial seizures. Seizure 5:259-265, 1996
- Beydoun A, Fischer J, Labar DR, et al: Gabapentin monotherapy: II. A 26-week, double-blind, dose-controlled, multicenter study of conversion from polytherapy in outpatients with refractory complex partial or secondarily generalized seizures. The US Gabapentin Study Group 82/83. Neurology 49:746-752, 1997
- Binnie D, Debets RM, Engelsman M, et al: Double-blind crossover trial of lamotrigine (Lamictal) as add-on therapy in intractable epilepsy. Epilepsy Res 4:222-229, 1989
- Biton V: Preliminary open-label experience with topiramate in primary generalized seizures. Epilepsia 38:S42-S44, 1997
- Blum DE: New drugs for persons with epilepsy. Adv Neurol 76:57-87, 1998
- Bourgeois B, Leppick IE, Sackellares JC, et al: Felbamate: A double-blind controlled trial in patients undergoing presurgical evaluation of partial seizures. Neurology 43:693-696, 1993
- Brodie MJ: Felbamate: A new antiepileptic drug. Lancet 341:1445-1446, 1993
- Brodie MJ, Dichter MA: Antiepileptic drugs. N Engl J Med 334:168-175, 1996
- Browne TR: Fosphenytoin (Cerebyx). Clin Neuropharmacol 20:1-12, 1997
- Butler WH: The neuropathology of vigabatrin. Epilepsia 30:S15-S17, 1989
- Callaghan N, Garrett A, Goggin T: Withdrawal of anticonvulsant drugs in patients free of seizures for 2 years. A prospective study. N Engl J Med 318:942-946, 1988
- Carter RB, Wood PL, Wieland S, et al: Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J Pharmacol Exp Ther 280:1284-1295, 1997
- Chadwick D: Gabapentin. Epilepsy Res Suppl 3:183-186, 1991
- Cheung H, Kamp D, Harris E: An in vitro investigation of the action of lamotrigine on neuronal voltage-activated sodium channels. Epilepsy Res 13:107-112, 1992
- Cramer JA, Mattson RH, Prevey ML, et al: How often is medication taken as prescribed? A novel assessment technique. JAMA 261:3273-3277, 1989
- Dailey JW, Seo DO, Yan QS, et al: The anticonvulsant effect of the broad spectrum anticonvulsant loreclezole may be mediated in part by serotonin in rats: A microdialysis study. Neurosci Letter 178:179-183, 1994
- Dam M, Ekberg R, Loyning Y, et al: A double-blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res 3:70-76, 1989
- Davies JA: Remacemide hydrochloride: A novel antiepileptic agent. Gen Pharmacol 28:499-502, 1997
- Devinsky O, Leppik I, Willmore LJ, et al: Safety of intravenous valproate. Ann Neurol 38:670-674, 1995
- Dooley JM, Camfield PR, Camfield CS, et al: Once-daily ethosuximide in the treatment of absence epilepsy. Pediatr Neurol 6:38-39, 1990
- Dulac O, Plouin P, Shewmon A: Myoclonus and epilepsy in childhood: 1996 Royaumont meeting. Epilepsy Res 30:91-106, 1998
- Faught E: Efficacy of topiramate as adjunctive therapy in refractory partial seizures: United States trial experience. Epilepsia 38:S24
- Faught E, Wilder BJ, Ramsay RE, et al. Topiramate placebo controlled dose ranging trial in refractory partial epilepsy using 200 , 400 , and 600 mg daily dosages. Topiramate YD Study Group. Neurology 46:1684 1690, 1996
- Ferrie CD, Robinson RO, Panayiotopoulos CP: Psychotic and severe behavioural reactions with vigabatrin: A review. Acta Neurol Scand 95:189 190, 1997
- Fisher RS, Kalviainen, Tanganelli P, et al: Newer antiepileptic drugs as monotherapy: Data on vigabatrin. Neurology 47:S2 S5, 1996
- French JA, Mosier M, Walker S, et al: A double blind, placebo controlled study of vigabatrin three g/day in patients with uncontrolled partial seizures. Vigabatrin Protocol 024 Investigative Cohort. Neurology 46:54 61, 1996
- Friedlander WJ: Who was the ‘father of bromide treatment of epilepsy’? Arch Neurol 43:505 507, 1986
- Friis ML, Kristensen O, Boas J, et al. Therapeutic experiences with 947 epileptic out patients in oxcarbazepine treatment. Acta Neurol Scand 87:224 227, 1993
- Garnett WR, Levy B, McLean AM, et al: Pharmacokinetic evaluation of twice daily extended release carbamazepine (CBZ) and four times daily immediate release CBZ in patients with epilepsy. Epilepsia 39:274 279, 1998
- Gram L: Tigabine: A novel drug with a GABAergic mechanism of action. Epilepsia 35:S85 S87, 1994
- Guerreiro MM, Vigonius U, Pohlmann H, et al: A double blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Res 27:205 213, 1997
- Handforth A, DeGiorgio CM, Schachter SC, et al: Vagus nerve stimulation therapy for partial onset seizures: A randomized active control trial. Neurology 51:48 55, 1998
- Harding GF: Severe persistent visual field constriction associated with vigabatrin. Benefit:risk ratio must be calculated for individual patients. BMJ 316:232 233, 1998
- Henry TR, Leppik IE, Gumnit RJ, et al: Progressive myoclonus epilepsy treated with zonisamide. Neurology 38:928 931, 1988
- Hill DR, Suman Chauhan N, Woodruff GN: Localization of [3H] gabapentin to a novel site in rat brain: Autoradiographic studies. Eur J Pharmacol 244:303 309, 1993
- Jawad S, Richens A, Goodwin G, et al: Controlled trial of lamotrigine (Lamictal) for refractory partial seizures. Epilepsia 30:356 363, 1989
- Kaufman DW, Kelly JP, Anderson T, et al: Evaluation of case reports of aplastic anemia among patients treated with felbamate. Epilepsia 38:1265 1269, 1997
- Kondo T, Kaneko S, Amano Y, et al: Preliminary report on teratogenic effects of zonisamide in the offspring of treated women with epilepsy. Epilepsia 37:1242 1244, 1996
- Krauss GL, Johnson MA, Miller NR: Vigabatrin associated retinal cone system dysfunction: Electroretinogram and ophthalmologic findings. Neurology 50:614 618, 1998
- Leppik IE, Willmore LJ, Homan RW, et al: Efficacy and safety of zonisamide: Results of a multicenter study. Epilepsy Res 14:165 173, 1993
- Loiseau P, Yuen AW, Duche B, et al: A randomized double blind placebo controlled cross over add on trial of lamotrigine in patients with treatment resistant partial seizures. Epilepsy Res 7:136 145, 1990
- Löscher W, Honack D, Rundfeldt C: Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther 284:474 479, 1998
- Luer MS: Fosphenytoin. Neurol Res 20:178 182, 1998
- Marson AG, Kadir ZA, Hutton JL, et al: The new antiepileptic drugs: A systematic review of their efficacy and tolerability. Epilepsia 38:859 880, 1997
- Matsuo F, Bergen D, Faught E, et al. Placebo controlled study of the efficacy and safety of lamotrigine in patients with partial seizures: US Lamotrigine Protocol No. 5 Clinical Trial Group. Neurology 43:2284 2291, 1993
- Mattson R, Cramer J, Collins J, et al: Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondary generalized tonic clonic seizures. N Engl J Med 313:145 151, 1985
- Mattson R, Cramer JA, Collins JF: A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondary generalized tonic clonic seizures in adults. The Department of Veterans Affairs Epilepsy Cooperative Study No. 264 Group. N Engl J Med 327:765 771, 1992
- Meldrum BS: Update on the mechanism of action of antiepileptic drugs. Epilepsia 37:S4 S11, 1996
- Merritt HH, Putnam TJ: A new series of anticonvulsant drugs tested by experiments on animals. Arch Neurol Psych 39:1003 1015, 1938
- Messenheimer J, Mullens EL, Giorgi L, et al: Safety review of adult clinical trial experience with lamotrigine. Drug Saf 18:281 296, 1998
- Messenheimer J, Ramsay RE, Willmore LJ, et al. Lamotrigine therapy for partial seizures: A multicenter, placebo controlled, double blind, cross over trial. Epilepsia 35:113 121, 1994
- Mikati MA, Holmes GL: Lamotrigine in absence and primary generalized epilepsies. J Child Neurol 12:S29 S37, 1997
- Monaghan EP, Navalta LA, Shum L, et al: Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia 38:1026 1031, 1997
- Muir KT, Palmer GC: Remacemide. Epilepsy Res Suppl 3:147 152, 1991
- Palhagen S, Canger R, Henriksen O, et al: Efficacy and safety of rufinamide in patients with refractory epilepsy. Epilepsia 38:3, 1997
- Pellock JM, Brodie MJ: Felbamate: 1997 update. Epilepsia 38:1261 1264, 1997
- Privitera M, Fincham R, Penry J, et al. Topiramate placebo controlled dose ranging trial in refractory partial epilepsy using 600 , 800 , and 1000 mg daily dosages. Neurology 46:1678 1683, 1996
- Ramsay RE, Wilder BJ, Uthman BM, et al: Intramuscular fosphenytoin (Cerebyx) in patients requiring a loading dose of phenytoin. Epilepsy Res 28:181 187, 1997
- Rentmeester T, Janssen A, Hulsman J, et al: A double blind, placebo controlled evaluation of the efficacy and safety of loreclezole as add on therapy in patients with uncontrolled partial seizures. Epilepsy Res 9:59 64, 1991
- Richens A, Chadwick DW, Duncan JS, et al. Adjunctive treatment of partial seizures with tiagabine: A placebo controlled trial. Epilepsy Res 21:37 42, 1995
- Rimmer EM, Richens A: Interaction between vigabatrin and phenytoin. Br J Clin Pharmacol 27:27 33, 1989
- Sachdeo RC, Leroy RF, Krauss GL, et al. Tigabine therapy for complex partial seizures. A dose frequency study. Tigabine Study Group. Arch Neurol 54:595 601, 1997
- Sander JW, Patsalos PN: An assessment of serum and red blood cell folate concentrations in patients with epilepsy on lamotrigine therapy. Epilepsy Res 13:89 92, 1992
- Sanna E, Murgia A, Casula A, et al: Direct activation of GABAA receptors by loreclezole, an anticonvulsant drug with selectivity for the beta subunit. Neuropharmacology 35:1753 1760, 1996
- Schachter SC: Tigabine monotherapy in the treatment of partial epilepsy. Epilepsia 36:S2 S6, 1995
- Schapel G, Chadwick D: Tigabine and non convulsive status epilepticus. Seizure 5:153 156, 1996
- Schapel GJ, Beran RG, Vajda FJ, et al. Double blind, placebo controlled, crossover study of lamotrigine in treatment of resistant partial seizures. J Neurol Neurosurg Psychiatry 56:448 453, 1993
- Schechter PJ, Hanke NF, Grove J, et al: Biochemical and clinical effects of gamma vinyl GABA in patients with epilepsy. Neurology 34:182 186, 1984
- Schmidt D, Jacob R, Loiseau P, et al: Zonisamide for add on treatment of refractory partial epilepsy: A European double blind trial. Epilepsy Res 15:67 73, 1993
- Seino M, Miyazaki H, Ito T: Zonisamide. Epilepsy Res Suppl 3:169 174, 1991
- Sharief M, Viteri C, Ben Menachem E, et al. Double blind, placebo controlled study of topiramate in patients with refractory partial epilepsy. Epilepsy Res 25:217 224, 1996
- Stefani A, Spadoni F, Bernardi G: Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacology 37:83 91, 1998
- Subramaniam S, Donevan SD, Rogawski MA: Block of the N methyl D aspartate receptor by remacemide and its des glycine metabolite. J Pharmacol Exp Ther 276:161 168, 1996
- Tassinari CA, Michelucci R, Chauvel P, et al. Double blind, placebo controlled trial of topiramate (600 mg daily) for the treatment of refractory partial epilepsy. Epilepsia 37:763 768, 1996
- The Tegretol OROS Osmotic Release Delivery System Study Group. Double blind crossover comparison of Tegretol XR and Tegretol in patients with epilepsy. Neurology 45:1703 1707, 1995
- The US Gabapentin Study Group No.5. Gabapentin as add on therapy in refractory partial epilepsy: A double blind, placebo controlled, parallel group study. Neurology 43:2292 2298, 1993
- UK Gabapentin Study Group. Gabapentin in partial epilepsy. Lancet 335:1114 1117, 1990
- Uthman BM, Rowan AJ, Ahmann PA, et al. Tiagabine for complex partial seizures. A randomized, add on, dose response trial. Arch Neurol 55:56 62, 1998
- Vigevano F, Cilio MR: Vigabatrin versus ACTH as first line treatment for infantile spasms: A randomized, prospective study. Epilepsia 38:1270 1274, 1997
- Wroblewski BA, Garvin WH, Jr. Once daily administration of phenobarbital in adults. Clinical efficacy and benefit. Arch Neurol 42:699 700, 1985